XFEL: New approach to imaging single biological particles
New approach to imaging single biological particles
As part of an international collaboration, scientists at European XFEL have developed and tested a novel approach for processing data from single biological particles such as proteins and viruses. Based on an idea first proposed over 40 years ago, the new method overcomes several problems of traditional approaches and could also have applications for other structural biology methods. The method is published today in the journal Physical Review Letters.
Solving the 3D structure of a biomolecule is often a rocky and windy journey. Traditionally, scientists have had to first force their molecule of choice to take on a crystalline form before shooting it with intense X-rays to reveal the 3D arrangement of the atoms. But not all biomolecules can be easily treated this way, and scientists can spend years trying to get their samples to crystallize. X-ray free-electron lasers (XFELs) such as the European XFEL now offer scientists with particularly stubborn samples a glimmer of hope. The increased brilliance and fast repetition rate of ultrashort X-ray flashes mean that it will be possible to take images of single particles, such as viruses and proteins, as they pass through the X-ray beam before the molecule itself is destroyed by the intense radiation—all without the need for extensive and time-consuming crystallization.
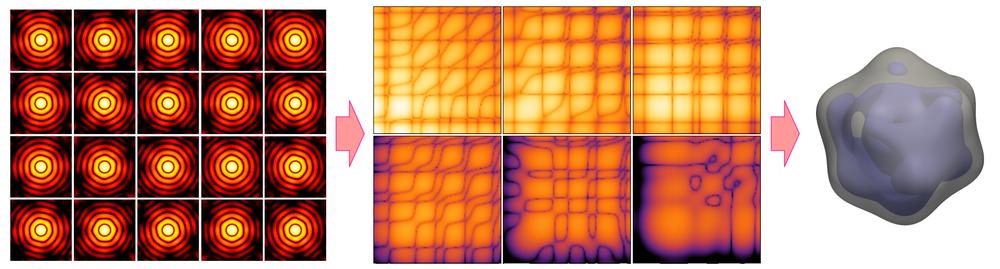
Schematic illustration of the new approach. Many X-ray diffraction snapshots recorded in the XFEL experiment (left) are reduced to a much more compact set of correlation data (centre), which act as a comprehensive 'fingerprint' of the 3D structure of a particle, such as a virus in this example. The correlation data are then used to reconstruct the 3D structure of a particle (right) by two analysis methods, model-based structure analysis and iterative phase retrieval by MTIP.
Step by step
In order to reconstruct a 3D structure of a molecule, conventional algorithms follow a number of consecutive steps. “If one step is not carried out correctly, this will have repercussions for the rest of the process,” explains Ruslan Kurta, theoretical physicist at European XFEL and lead author on today’s paper. “Each of these steps can be tricky,” he adds. “The good news, however, is we have now developed and successfully tested an approach that addresses some of the challenges in this process,” says Adrian Mancuso, co-author and lead scientist for the single-particle imaging and serial crystallography instrument (SPB/SFX) of the European XFEL.

European XFEL theoretical physcist Ruslan Kurta is the lead author on the paper.
A major hurdle in most single-particle analysis methods is that of orientation determination. “In a single-particle experiment, one does not have control over rotation of the particles as they are hit by the X-ray beam, and consequently each snapshot from a successful hit will contain information about the sample from a different random orientation,” explains co-author Jeff Donatelli, a member of Berkeley Lab’s Center for Advanced Mathematics for Energy Research Applications (CAMERA) who developed many of the algorithms in the new method. “Most approaches to single-particle analysis have so far been based on trying to determine these particle orientations from the images; however, the best resolution achievable from these analyses is restricted by how precisely these orientations can be determined from the data,” he adds. Get this wrong, and the following data processing steps will also include errors.
The new approach developed by the group circumvents this problem and fulfills the promise of an idea first proposed 40 years ago. “Rather than examine the individual data intensities in an attempt to find the correct orientation for each measured frame, we eliminate this step altogether by using so-called cross-correlation functions,” explains Kurta. ”The beauty of using correlation data is that it contains a comprehensive fingerprint of the 3D structure of an object,” explains co-author Peter Zwart, also a member of CAMERA at Berkeley Lab.
Since the idea of using correlation functions was first proposed in the 1970s, several research groups have tried to use them to overcome the problem of orientation determination. First experiments to test the feasibility of using correlation functions were done at DESY and EMBL in Hamburg in the 1980s. “However, the X-ray sources available back then were just not bright enough to get enough data to show that the idea could work,” says Zwart. “But now, 40 years later, technology has caught up and we have all the resources in place to finally make those original ideas a reality!” he adds.
Doing away with symmetry constraints
Another issue is that other approaches have all relied on symmetry assumptions of the investigated particle. “The problem with this,” says Donatelli, “is that these assumptions are not always applicable to biological particles and may also prevent us from detecting biologically relevant asymmetries in our samples.” “That means, for example, that we would assume a virus is a perfectly, symmetrical shaped capsid. But,” Kurta adds with a smile, “If you put symmetry in, you’ll get symmetry out!” Kurta and colleagues have managed to do away with the need for any symmetry assumptions making it possible to observe additional features of the molecule shape. The team tested their approach by collecting data on several nanoscale viruses at the AMO beamline of LCLS at SLAC in the USA. “We were really quite amazed to see that, in contrast to previously published structural data of the same virus, the virus particles were actually not symmetrical” Kurta says. Just why the viruses are not symmetrical, they can’t say. “Maybe there really is some natural variation in shape and density, or possibly the asymmetry is induced by the specific experimental conditions” says Kurta. “These possibilities are all feasible, as shown by our modelling data, and although we can’t be sure what the real reason is, it’s important that we can observe these differences at all”. The results open up important discussions about how such experiments are designed, how the sample is moved into the X-ray beam and how to best understand the structure of single viruses.
Recovering 3D structures
A key component of the team’s breakthrough is the so-called multi-tiered iterative phasing (M-TIP) algorithm developed by CAMERA, designed to achieve the ultimate goal of determining the 3D structure of the molecules. “Among the prominent advantages of M-TIP is its ability to solve the structure directly from the correlation data without having to rely on any symmetry constraints, and, more importantly, without the need to solve the orientation determination problem,” explains Donatelli. “Advanced correlation analyses in combination with reconstructions by M-TIP clearly define an efficient route for structural analysis of nanoscale objects at XFELs,” explains Zwart.
More data for better resolution
The international team is not satisfied yet, however, and plan more experiments to further improve the method. “Currently, all single-particle imaging experiments suffer from a limitation in the number of data frames collected,” explains Mancuso. “The more data you can collect, the better the signal and consequently the resolution achievable.” This will be particularly important as scientists attempt to solve the structures of increasingly smaller molecules. “Most algorithms for single-particle imaging can only handle one molecule at a time, thus limiting signal and resolution. Our approach, on the other hand, is scalable so that we should also be able to measure more than one particle at a time,” says Kurta.
“With more good data we’d have a good chance of improving the resolution and robustness of single-particle imaging methods,” says Mancuso excitedly, “and with their higher repetition rate, facilities such as the European XFEL, will allow us to collect much more good data quickly—potentially improving our 3D structure determinations with correlation methods and more.”
With these characteristics, the team also sees potential for the use of their approach beyond the field of single-particle imaging. “Other structural biology techniques such as SAXS measure the structure of molecules suspended in solution, and processing of SAXS data faces some of the same challenges faced by single-particle imaging,” explains Kurta. “Since our approach can measure more than one particle at a time, it may also help with improving SAXS data processing methods at X-ray radiation sources such as synchrotrons as they are upgraded to produce more intense X-ray beams.”
“We are really excited about this approach!” says Mancuso. “Together with the XFEL technological advances, such as those we are currently seeing at European XFEL, we believe we can really push the limits of what is possible for exploring the structure of biological molecules.”
Original publication:
Correlations in Scattered X-Ray Laser Pulses Reveal Nanoscale Structural
Features of Viruses
Kurta et al., Phys. Rev. Lett. published online 12 October 2017
DOI: 10.1103/PhysRevLett.119.158102
